Applying machine learning to guide ensemble curation¶
An ensemble of models can be though of as a set of feasible hypotheses about how a system behaves. From a machine learning perspective, these hypotheses can alternatively be viewed as samples (or observations), each of which has a distinct set of features (i.e. the model components that vary across an ensemble) and can further generate new features by performing simulations. An example of the analyses enabled by this view of ensembles can be found in Medlock & Papin, where ensemble structure and ensemble simulations are used to identify reactions that are high-priority targets for curation.
In this example, we demonstrate how ensembles of genome-scale metabolic models and machine learning can be combined to identify reactions that might strongly influence a single prediction (flux through biomass). We will use an ensemble for Staphylococcus aureus that contains 1000 members. The ensemble was generated through iterative gapfilling to enable growth on single C/N media conditions using a draft reconstruction from ModelSEED.
In [1]:
import medusa
from medusa.test import create_test_ensemble
ensemble = create_test_ensemble("Staphylococcus aureus")
Using the ensemble, we’ll perform flux balance analysis and return flux
through the biomass reaction (which has an ID of "bio1"). The
ensemble already has the media conditions set as “complete”, meaning the
boundary reactions for all transportable metabolites are open (e.g. the
lower bound of all exchange reactions is -1000).
In [2]:
from medusa.flux_analysis import flux_balance
biomass_fluxes = flux_balance.optimize_ensemble(ensemble, return_flux="bio1", num_processes = 4)
The optimize_ensemble function returns a pandas DataFrame, where
each column is a reaction and each row is an ensemble member. For
illustration, here are the values for the first 10 members of the
ensemble:
In [3]:
biomass_fluxes.head(10)
Out[3]:
| bio1 | |
|---|---|
| Staphylococcus aureus_gapfilled_518 | 118.238182 |
| Staphylococcus aureus_gapfilled_860 | 122.523063 |
| Staphylococcus aureus_gapfilled_900 | 104.905551 |
| Staphylococcus aureus_gapfilled_434 | 148.353976 |
| Staphylococcus aureus_gapfilled_343 | 134.100850 |
| Staphylococcus aureus_gapfilled_706 | 116.982207 |
| Staphylococcus aureus_gapfilled_175 | 137.352545 |
| Staphylococcus aureus_gapfilled_85 | 110.488964 |
| Staphylococcus aureus_gapfilled_345 | 119.439103 |
| Staphylococcus aureus_gapfilled_161 | 118.237318 |
To get a sense for the distribution of biomass flux predictions, we can visualize them with matplotlib:
In [4]:
import matplotlib.pylab as plt
fig, ax = plt.subplots()
plt.hist(biomass_fluxes['bio1'], bins = 15, color = 'black', alpha = 0.4)
ax.set_ylabel('# ensemble members')
ax.set_xlabel('Flux through biomass reaction')
plt.savefig('pre_FBA_curation.svg')
plt.show()
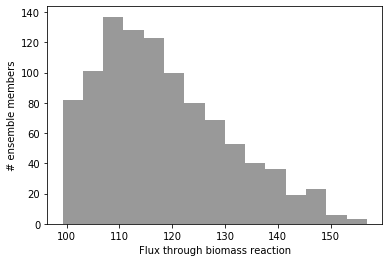
As you can see, there is quite a bit of variation in the maximum flux through biomass! Keep in mind that this is an ensemble of gapfilled reconstructions with no manual curation, and that none of the uptake rates are reallistically constrained, so these predictions are unrealistically high (100 units of flux through biomass is a doubling time of 36 seconds, at least an order of magnitude faster than even the fittest E. coli grown in vitro).
Our goal now is to identify which features in the ensemble are predictive of flux through biomass. If we can identify these reactions, then turn to the literature or perform an experiment to figure out whether they are really catalyzed by the organism, we can greatly reduce the uncertainty in our predictions of biomass flux!
Given that we have a continous output, our problem can be addressed using regression. We will use the binary presence/absence of each reaction in each member of the ensemble as input to a random forest regressor, implemented in scikit-learn. Many supervised regression models will work for this analysis, but random forest is particularly easy to understand and interpret when the input is binary (i.e. reaction presence/absence).
In [5]:
import sklearn
from sklearn.ensemble import RandomForestRegressor
We reformat the data here, getting the feature states for each ensemble
member and converting them to True/False, then combine them into
a single DataFrame with the biomass flux predictions for matched
members.
In [6]:
# Grab the features and states for the ensemble and convert to a dataframe
import pandas as pd
feature_dict = {}
for feature in ensemble.features:
feature_dict[feature.id] = feature.states
feature_frame = pd.DataFrame.from_dict(feature_dict)
# convert the presence and absence of features to a boolean value
feature_frame = feature_frame.astype(bool)
# extract biomass and add it to the dataframe, keeping track of the feature names
input_cols = feature_frame.columns
biomass_fluxes.index = [member_id for member_id in biomass_fluxes.index]
feature_frame['bio1'] = biomass_fluxes['bio1']
Now we actually construct and fit the random forest regressor, using 100
total trees in the forest. The oob_score_ reported here is the
coefficient of determination (R2) calculated using the out-of-bag
samples for each tree. As a reminder, R2 varies from 0 to 1.0, where 1.0
is a perfect fit.
In [7]:
# create a regressor to predict biomass flux from reaction presence/absence
regressor = RandomForestRegressor(n_estimators=1000,oob_score=True)
fit_regressor = regressor.fit(X=feature_frame[input_cols],y=feature_frame['bio1'])
fit_regressor.oob_score_
Out[7]:
0.8684706250117109
With a reasonably-performing regressor in hand, we can inspect the important features to identify reactions that contribute to uncertainty in biomass flux predictions.
In [8]:
imp_frame = pd.DataFrame(fit_regressor.feature_importances_,
index=feature_frame[input_cols].columns).sort_values(
by=0,ascending=False)
imp_frame.columns = ['importance']
In [9]:
imp_frame.head(10)
Out[9]:
| importance | |
|---|---|
| rxn01640_c_upper_bound | 0.129785 |
| rxn01640_c_lower_bound | 0.113434 |
| rxn12585_c_lower_bound | 0.044854 |
| rxn12585_c_upper_bound | 0.042830 |
| rxn15617_c_lower_bound | 0.039388 |
| rxn23244_c_lower_bound | 0.039336 |
| rxn00602_c_lower_bound | 0.037124 |
| rxn15617_c_upper_bound | 0.036253 |
| rxn00602_c_upper_bound | 0.032443 |
| rxn23244_c_upper_bound | 0.028802 |
With the list of important features in hand, the first thing we should
do is turn to the literature to see if someone else has already figured
out whether these reactions are present or absent in Staphylococcus
aureus. The top reaction, rxn01640, is N-Formimino-L-glutamate
iminohydrolase, which is part of the histidine utilization pathway. A
quick consultation with a review on the regulation of histidine
utilization in bacteria
suggests that the enzyme for this reaction, encoded by the hutF gene,
is widespread and conserved amongst bacteria. However, the hutF gene
is part of a second, less common pathway that branches off of the
primary histidine utilization pathway. If we consult PATRIC with a
search for the *hutF*
gene, we
see that, although the gene is widespread, there is no predicted hutF
gene in any sequenced Staphylococcus aureus genome. Although absence
of evidence is not evidence of absence, we can be relatively confident
that hutF is not encoded in the Staphylococcus aureus genome, given
how well-studied this pathogen is.
What happens if we “correct” this issue in the ensemble? Let’s inactivate the lower and upper bound for the reaction in all the members, then perform flux balance analysis again.
In [10]:
for member in ensemble.features.get_by_id('rxn01640_c_lower_bound').states:
ensemble.features.get_by_id('rxn01640_c_lower_bound').states[member] = 0
ensemble.features.get_by_id('rxn01640_c_upper_bound').states[member] = 0
biomass_fluxes_post_curation = flux_balance.optimize_ensemble(ensemble, return_flux="bio1", num_processes = 4)
In [11]:
import matplotlib.pylab as plt
import numpy as np
fig, ax = plt.subplots()
# declare specific bins for our histogram
bins=np.histogram(np.hstack((biomass_fluxes['bio1'],biomass_fluxes_post_curation['bio1'])),
bins=15)[1]
plt.hist(biomass_fluxes['bio1'],
bins,
label = 'Original',
alpha = 0.3,
color='black')
plt.hist(biomass_fluxes_post_curation['bio1'],
bins,
label = 'Post-curation',
alpha = 0.4,
color = 'green')
plt.axvline(x=biomass_fluxes['bio1'].mean(), c = 'black', alpha = 0.6)
plt.axvline(x=biomass_fluxes_post_curation['bio1'].mean(), c = 'green')
ax.set_ylabel('# ensemble members')
ax.set_xlabel('Flux through biomass reaction')
plt.legend(loc='upper right')
plt.savefig('post_FBA_curation.svg')
plt.savefig('post_FBA_curation.png')
plt.show()
In [ ]:
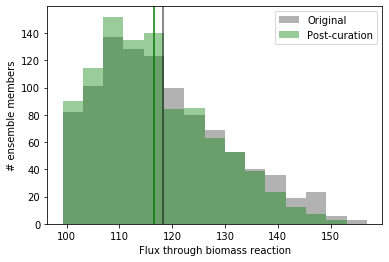
Here, we show the old distribution in gray and the new distribution in
green, with vertical lines at the mean in the same color. As you can
see, by resolving the identity of the hutF-encoded enzyme, we’ve
reduced the mean and range of predicted flux through biomass. The
reduction here is modest, but the process can be repeated for the other
important features we identified to continue to refine the distribution
and improve the reconstruction in a rational way.